Choose input mode
|
Mode 1: Submit a query protein (execute Steps 1 - 4 of the Mustguseal protocol)
Mode 2: Submit a core structural alignment (execute Steps 3 - 4 of the Mustguseal protocol)
Mode 3: Submit a core structural alignment and results of sequence similarity search (execute Step 4 of the Mustguseal protocol)
Mode 4 (Mustguseal + Zebra3D): Submit a query protein to construct only the core 3D-structural alignment and run Zebra3D analysis
Claim your results by TaskID (access the results and progress log of a previously submitted task)
Select one of the four input modes to submit a new task or access a previously submitted task
|
Choose a scenario
Scenarios can be used in Modes 1 and 2 to instantly load a pre-selected collection of parameters to construct a multiple protein alignment for a particular purpose. Scenarios can be very helpfull at creating high-quality content-rich output and make your experience with Mustguseal more comfortable and efficient. Please have a look at the available scenarios and select the one which seems to be closer to your objectives.
Scenario 1: "The default"The default parameters are used as explained in the Mustguseal publication Suplatov et al. (2018) Bioinformatics.Scenario 2: "Alignment of a large representative set of functionally diverse proteins with high structural but low sequence similarity to the query for further analysis by Zebra, pocketZebra, and visualCMAT"This scenario can be used to construct an alignment for further analysis by Zebra, pocketZebra, and visualCMAT web-servers. The Mustguseal will automatically collect a large representative set of functionally diverse proteins from public databases with high structural but low sequence similarity to the query (in the Mode 1). If the submission is made in the Mode 2, then the level of structural similarity in the final output alignment will be defined by the level of structural similarity in the user-submitted core structural alignment. The Scenario 2 is based on Scenario 1 ("The default") with the following changes: the UniProtKB/Swiss-Prot+TrEMBL databases are used; the Redundancy filter is set to 90%; the Sequence length filter is disabled by setting the threshold to 9999%; the Use MAPU to build large alignments is set to Yes.Scenario 3: "Alignment of a complete set (i.e., redundancy filter set to 100%) of functionally diverse proteins with high structural but low sequence similarity to the query for further analysis by Yosshi"This scenario can be used to construct an alignment for further analysis by Yosshi web-server. It is based on the scenario 2 but the redundancy filter is set to 100% to remove only identical sequences and the sequence length filter is not applied, i.e., the length of proteins which are collected by the sequence similarity search is not limited to the length of corresponding representative proteins. This can result in the increased number of alignment columns with a high content of gaps, which is not a problem for Yosshi as that web-server dismisses the gaps and considers all amino acids residues in all columns for a potential disulfide connectivity. Trimming of the alignment by the sequences of representative proteins (i.e., use of the MAPU tool) is turned on to decrease the size of the output alignment. The Scenario 3 is based on Scenario 1 ("The default") with the following changes: the UniProtKB/Swiss-Prot+TrEMBL databases are used; the Maximum number of sequences in each subsearch is set to 1000; the Redundancy filter is set to 100%; the Sequence length filter is disabled by setting the threshold to 9999%; the Use MAPU to build large alignments is set to Yes. |
The parameters have been loaded according to the chosen scenario.
Please note that you might need to adjust these parameters depending on your research objectives and specific structural/domain organization
of proteins in your superfamily. The guidelines for parameters selection and examples are provided in the
Supplementary Data to
Suplatov D.A. et al. (2018) Bioinformatics 10.1093/bioinformatics/btx831.
Please note that changing the input Mode in the box above will reload the parameters to Scenario 1 "The default".
Submit a new task in Mode 1
Parameters setup |
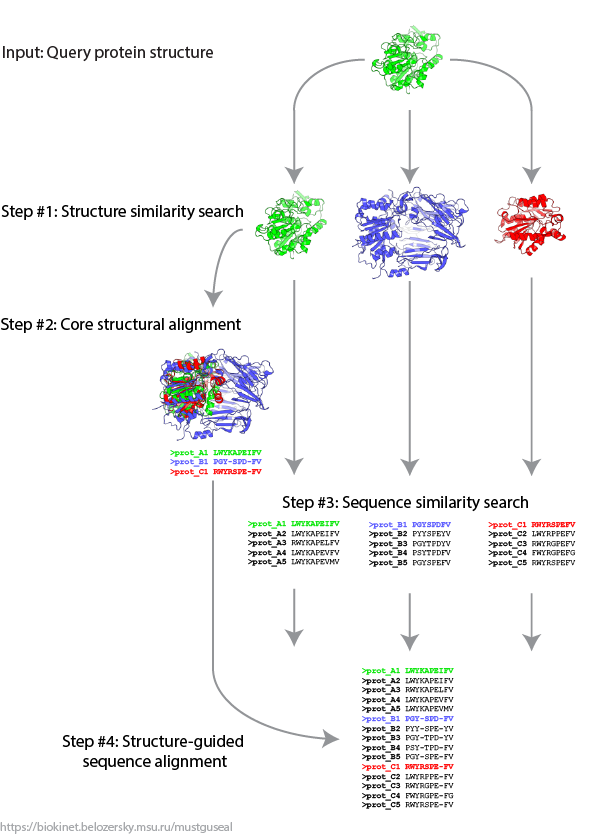
Algorithm outline |
||||||||||||||||||||
|
|
|
||||||||||||||||||||